Research

Comparison of genetic diversity across the paleopolyploid genome of Brassica rapa. Genomic data from more than 100 newly sequenced accessions of B. rapa crops are included in the analysis. These include turnips, field mustards, Chinese cabbage, pak choi, sarsons, and rapini. From Qi et al. 2021 New Phytologist
We study the origins of biological diversity, particularly how abrupt genomic events such as polyploidy (genome duplication), chromosomal change, and hybridization have contributed to the evolution and diversity of life. Biologists have long been fascinated by these processes because they create unique opportunities for the evolution of novelty with the potential for relatively rapid diversification. While assessing the roles of these genomic changes in evolution has historically been a difficult task, advances in genomics and computational biology have created new opportunities for addressing these longstanding questions.
Our research program integrates new computational and evolutionary genomic tools with traditional approaches such as molecular evolution, phylogenetics, mathematical modeling, field collections, and experimental work to better understand the origins of biological diversity. Prospective students and postdocs can engage with any combination of these approaches depending on their interests and career goals. We use a combination of publicly available genomic data and new data generated by ourselves and collaborators from diverse study systems spanning the Sonoran Desert and Madrean Sky Islands to global phylogenomic datasets. Our ultimate goal is to connect patterns of genome evolution across time scales by leveraging systems where we can study microevolutionary processes to inform our understanding of macroevolutionary patterns.

Simulation analyses of the diversification of diploid and polyploid lineages with independent speciation and extinction rates. From Arrigo & Barker 2012.
Macroevolutionary Patterns and Diversification
How does polyploidy influence large-scale patterns of speciation, extinction, and biodiversity accumulation across evolutionary time?
Polyploidy, both recent and ancient, is common throughout plant evolution and shapes diversification patterns across lineages. In the Brassicaceae, polyploid lineages show both higher speciation and extinction rates compared to diploids, yet contribute to greater net diversity over evolutionary time (Román-Palacios et al. 2020, Proceedings B). Although polyploid speciation occurs frequently (Wood et al. 2009, PNAS), newly formed polyploids often exhibit lower initial diversification than their diploid relatives (Mayrose et al. 2011, Science). Despite leaving a substantial legacy in plant genomes, the long-term evolutionary success of most polyploid species appears constrained, with elevated extinction rates suggesting that only a subset of polyploid lineages persist and contribute to the diversity of plants (Arrigo & Barker 2012, Current Opinion). The interplay between polyploidy and chromosome number changes (dysploidy) adds further complexity, with variation in these processes correlating with clade-specific diversification patterns (Zhan et al. 2021, bioRxiv). Understanding these multifaceted relationships (Landis et al. 2018, AJB) requires integrating data across timescales from contemporary populations to deep phylogenetic patterns.
Key Findings:
Higher speciation AND extinction in polyploid species
Net diversity increase despite turnover
Variable patterns across lineages
Ancient WGDs shape modern diversity
Methods:
Trait-dependent diversification models
Comprehensive ploidy inference
Fossil-calibrated phylogenies
Integrated diversification analyses
Open Questions:
What determines polyploid success versus failure?
How do environmental changes affect polyploid diversification?
Can we predict rapid diversification potential?
How do WGDs interact with other drivers of biodiversity?
Opportunities for: macroevolutionary modeling, diversification analyses, fossil calibration work, and biodiversity synthesis

The number of rounds of WGDs inferred in the history flowering plant species. After McKibben et al. 2024 American Journal of Botany.
Phylogenomic Approaches to WGD Evolution
How can we accurately detect and characterize ancient genome duplications across the tree of life? What do large-scale phylogenomic analyses reveal about the timing, frequency, and evolutionary consequences of WGDs?
To understand the broader evolutionary significance of genome duplications, we analyzed WGD patterns across the green plant phylogeny as part of the One Thousand Plant Transcriptomes Initiative (2019, Nature). We found evidence for nearly 250 ancient WGDs across plants with multiple rounds of WGD characterizing many land plant lineages. More recent research demonstrated that phylogenetic uncertainty significantly impacts WGD inference (McKibben et al. 2024, American Journal of Botany). We have uncovered ancient duplications in conifers (Li et al. 2015, Science Advances), parallel paralog retention patterns (Barker et al. 2008, MBE) across nested WGDs in the Compositae (Barker et al. 2016, AJB), and multiple WGDs across the Brassicales (Mabry et al. 2020, AJB). Our analyses suggest genome duplications cluster at key evolutionary transitions and environmental changes rather than occurring randomly.
Key Findings:
Complex duplication histories across land plants
Phylogenetic uncertainty affects WGD inference
Non-random WGD distribution across clades
Multiple genomic features improve detection accuracy
Methods:
Large-scale phylogenomic analysis
Uncertainty-aware WGD inference
Synteny and gene tree integration
Machine learning detection algorithms (SLEDGE)
Open Questions:
How do we handle phylogenetic uncertainty in WGD studies?
What determines ancient WGD detectability?
Can we robustly date WGDs with rate variation?
What drives genome duplication timing?
Opportunities for: large-scale data analysis, phylogenetic methods, computational biology, and collaborative projects with international consortia

A riparian plot showing genome evolution across five angiosperm species with different rounds of WGD. McKibben and Barker unpublished.
Diploidization and Post-Polyploid Genome Evolution
Why do polyploid species diploidize if they were successful as polyploids? Does natural selection or drift drive diploidization? Are there universal rules that govern genome evolution following genome duplication?
Our analyses of diploidization processes reveals that most contemporary plant species are ancient polyploids that underwent extensive genomic reorganization (Li et al. 2021, Annual Review of Plant Biology). We have found that diploidization generally involves three different processes: genetic diploidization (restoration of disomic inheritance), cytological evolution (typically reduction in chromosome numbers), and gene fractionation (duplicate gene loss). Recent work comparing ferns and angiosperms shows they have similar rates of ancient polyploidy, but ferns retain chromosomes much longer (Li et al. 2024, bioRxiv). These results reveal lineage-specific patterns of diploidization and post-polyploid genome evolution that can be used to understand the biology of diploidization.
Key Findings:
Variable diploidization rates across lineages create genomic diversity
Different processes of diploidization and post-polyploid genome evolution appear to proceed independently
Gene retention shows both stochastic and selective patterns
Synteny conservation varies dramatically between plant groups
Methods:
Comparative fractionation analysis
Machine learning classification of WGD paralogs (Frackify)
Synteny-based analyses (SynTRACE)
Phylogenomic integration approaches
Open Questions:
What determines diploidization rates across lineages?
Can we predict gene retention following duplication?
How do environmental factors influence post-WGD evolution?
What maintains synteny conservation in some lineages?
Opportunities for: computational phylogenomics, comparative genomics, and collaborative projects with plant genome consortia
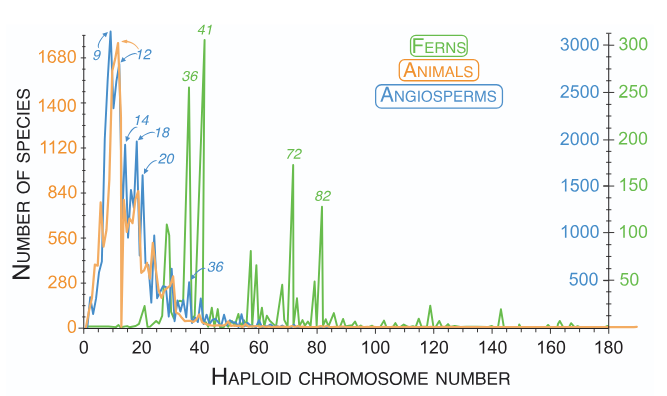
The distribution of chromosome numbers among flowering plants, ferns, and animals. After Román-Palacios et al. 2021
Chromosome Number Evolution
Why do rates of chromosome gain and loss vary dramatically across the phylogeny? What evolutionary forces drive the gain and loss of chromosomes, and ultimately the evolution of chromosome number?
Plants and animals show remarkably similar chromosome number ranges despite different evolutionary histories, suggesting universal constraints on genome organization (Román-Palacios et al. 2021, Journal of Evolutionary Biology). One recent hypothesis proposed that selection to reduce chromosome numbers is caused by the the number of segregating RGL sets in a genome, but our research has found no correlation between chromosome number reproductive isolation across 94 plant genera (Finch et al., in prep). Recent research on meiotic drive provides a different mechanistic insight: differences in meiotic symmetry between heterosporous and homosporous plants may drive their distinct patterns of chromosome number evolution (Plačková et al. 2024; Kinosian & Barker 2025, Molecular Ecology).
Key Findings:
Meiotic drive profoundly impacts chromosome evolution
High numbers of chromosomes in ferns and the rapid loss of chromosomes in angiosperms may both be explained by drive
Annual plants show 11.41x higher chromosome loss rates
No correlation between chromosome number and reproductive barriers
Methods:
Comparative karyotype analysis
Analyses to test for the signatures of meiotic drive
Extreme systems (Xanthisma n = 2) - see Study Systems
ChromEvol and BiChroM modeling
Open Questions:
How does meiotic symmetry influence chromosome evolution?
What enables extreme chromosome reduction?
Can meiotic drive explain major plant differences?
How do life history traits predict genome trajectories?
Opportunities for: field collections, cytological work, theoretical modeling, and comparative analyses
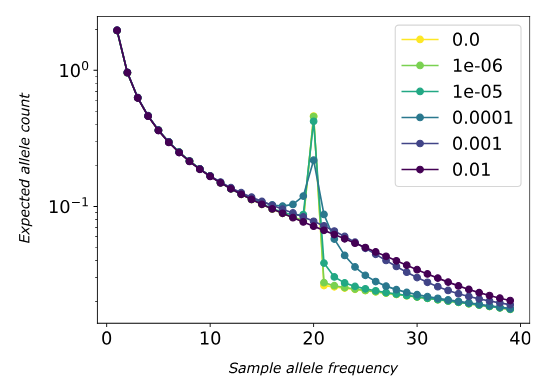
Effect of homoeologous exchange rate on the tetraploid site frequency spectrum. High exchange rates eliminate the characteristic 50% frequency peak as alleles mix, causing the SFS to resemble that of an autopolyploid. After Blischak et al. 2023, Genetics
Population Genomics of Polyploid Systems
How do multiple origins of polyploid species contribute to polyploid success? How do the population genetics of polyploid species influence their macroevolutionary outcomes?
We develop theoretical frameworks for polyploid population processes, addressing the unique challenges of complex inheritance patterns and genomic architectures. Our work reveals autopolyploids are far more common than assumed, challenging the allopolyploid-dominated view (Barker et al. 2016, New Phytologist). Through collaborations with the Gutenkunst lab in MCB, we have developed new statistical methods to study polyploid population genetics (Blischak et al. 2020, Molecular Biology & Evolution; Blischak et al. 2023, Genetics). These new population genetic tools are crucial for understanding polyploid establishment and long-term evolutionary fate.
Key Findings:
Complex inheritance creates unique evolutionary trajectories
Multiple origins common in polyploid formation
Homoeolog exchange affects selection efficacy
Demographic patterns differ from diploid expectations
Methods:
Polyploid demographic modeling
Low-coverage SNP calling methods
Inheritance pattern estimation
Complex mating system analysis
Open Questions:
Do polyploids arise via bottlenecks or multiple origins?
How do inheritance patterns affect evolutionary fate?
Can we predict polyploid establishment success?
How do micro- and macroevolution connect?
Opportunities for: population genetics theory, statistical method development, field sampling, and genomic data analysis
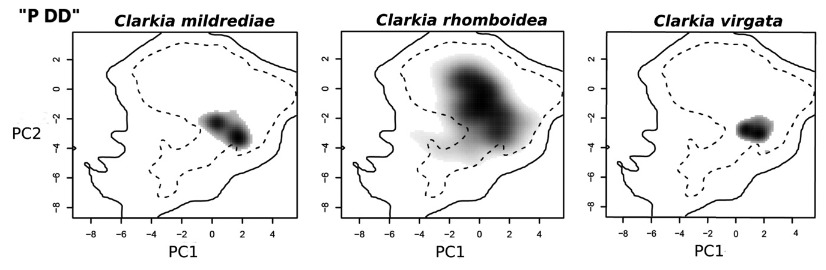
Climatic niche overlap between an allopolyploid and its diploid progenitors. Grey shading shows species occurrence density; contour lines indicate 100% (solid) and 50% (dashed) of available environmental space. This example shows an allopolyploid with less climatic overlap with both parents than the parents share with each other. After Baniaga et al. 2020, Ecology Letters
Climate Adaptation and Ecological Genomics
How does polyploidy influence rates of adaptation and speciation? Do polyploid species demonstrate greater ecological plasticity than their diploid relatives?
Polyploids evolve into new ecological niches significantly faster than diploids, supporting the hypothesis that genome duplication enhances adaptive flexibility (Baniaga et al. 2020, Ecology Letters). Gene duplications also drive co-evolutionary dynamics, as shown in butterfly-plant chemical defense arms races where successive duplications enabled novel defense evolution (Edger et al. 2015, PNAS). These enhanced adaptive capacities appear especially critical in the context of rapid environmental change, where genomic redundancy provides both mutational buffering and the evolutionary substrate needed for rapid adaptation. By studying these mechanisms, our research directly connects fundamental evolutionary processes to the capacity of species to respond to climate change.
Key Findings:
Accelerated multivariate niche evolution in polyploids
Genome duplication facilitates novel niche occupation
Enhanced stress response potential
Co-evolutionary advantages through gene duplication
Methods:
Ecological niche modeling
Environmental gradient sampling
Comparative phylogenetic approaches
Genomic-ecological data integration
Open Questions:
What mechanisms link polyploidy to ecological flexibility?
How do polyploids respond to rapid environmental change?
Can we predict climate resilience from genome structure?
Which genomic features confer adaptive advantages?
Opportunities for: field work in the Sonoran Desert and Sky Islands, ecological modeling, conservation genomics, and climate change research
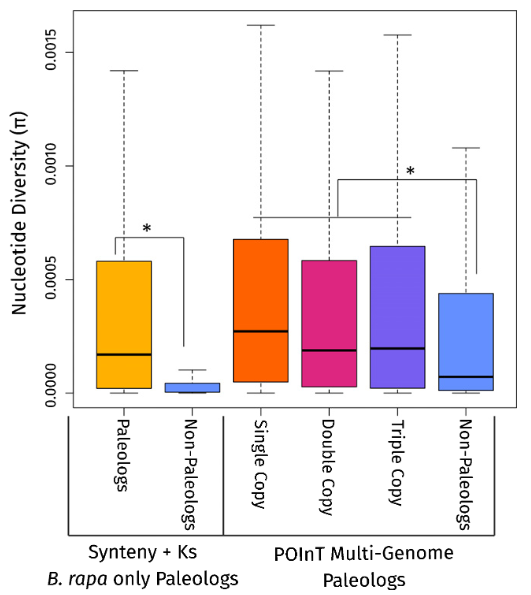
Comparison of nucleotide diversity (p) across Brassica rapa
paleologs and non-paleologs. After Qi et al. 2021, New Phytologist
Agricultural Genomics and Crop Domestication
How do ancient genome duplications continue to influence contemporary agriculture? Can evolutionary genomics inform crop improvement strategies?
Polyploidy appears to be a fundamental driver of crop domestication success, with ancient genome duplications providing the genetic foundation for agricultural innovation. Our analyses across 22 crop species reveals that paleologs (ancient duplicate genes) are consistently overrepresented in domestication genes, linking paleopolyploidy to agricultural success (McKibben and Barker, in prep). In Brassica rapa, paleologs harbor more genetic diversity and are enriched among candidate gene lists (Qi et al. 2021, New Phytologist). The legacy of variation from ancient WGDs provided the raw material for artificial selection millions of years later. Understanding this evolutionary foundation offers strategies for climate-adapted crop development.
Key Findings:
Paleolog enrichment in domestication genes across many crops
Higher genetic diversity in paralogs retained from ancient WGDs
Single-copy paleologs most enriched
Ancient WGDs enable modern crop improvement
Methods:
Machine learning gene classification (Frackify)
Population genomics of crop diversity
Phylogenetic comparative approaches
Selection analyses
Open Questions:
How do paleologs contribute to specific traits?
Can evolutionary insights guide crop improvement?
Which paleologs predict breeding success?
How does selection act differently on duplicated genes?
Opportunities for: computational genomics, agricultural collaborations, molecular evolution analyses, and applied research with societal impact
Bioinformatic Tool Development
How can machine learning and computational approaches detect complex genomic patterns that traditional methods cannot capture?
We develop computational tools that uncover previously inaccessible patterns in genome evolution. Our machine learning frameworks address previously intractable problems from gene classification to reconstruction of synteny networks. These computational tools enable analyses that were previously impractical—integrating heterogeneous datasets, testing complex multivariate hypotheses, and scaling analyses from single genes to entire genomes.
Key Tools:
Frackify: Gradient boosted trees for gene classification
HyDe-CNN: Neural networks for hybrid detection
SynTRACE: Synteny-based genome reconstruction
Ploidify: Probabilistic ploidy inference
GOgetter & GOmosaic: Polyploid functional annotation
Open Questions:
How can we improve evolutionary inference accuracy?
What patterns emerge from machine learning approaches?
Can we predict post-duplication evolutionary outcomes?
How do we best integrate diverse genomic evidence?
Opportunities for: machine learning, software development, open-source contributions, and computational methods training
Joining Our Research Program
If you are interested in exploring these evolutionary questions about genome duplications, polyploidy, and biodiversity, we welcome you to join our lab. Please visit our Contact page for more information about current opportunities and how to apply.